代谢是细胞内的各种化学反应的总称,用于合成或分解大分子和小分子化合物。这些化合物很多是多肽、脂质、有机酸等,除了维持细胞的营养和能量供应,在信号转导、细胞通讯和转录调控方面都有重要作用。
麻烦的是,代谢物的类别太复杂,现在还没有哪种方法可以一次性把细胞里所有的代谢物都测出来。但另一个方面,代谢反应大多需要酶做催化,酶就是基因的表达产物,而基因表达的转录组是可以测的。那么从基因表达如何推导出代谢物丰度,有了代谢物以后又会带来什么新发现?本文介绍一种使用流平衡分析(flux balance analysis)来做细胞代谢强度推断的方法,再介绍一种用代谢物-受体配对数据库来做细胞通讯的方法。
Compass:FBA推断单细胞的代谢强度
第一篇文章是Metabolic modeling of single Th17 cells reveals regulators of autoimmunity,2021年8月发表在Cell。作者是Nir Yosef,也是scVI的作者,来自UC Berkeley的团队,Aviv Regev也在作者里
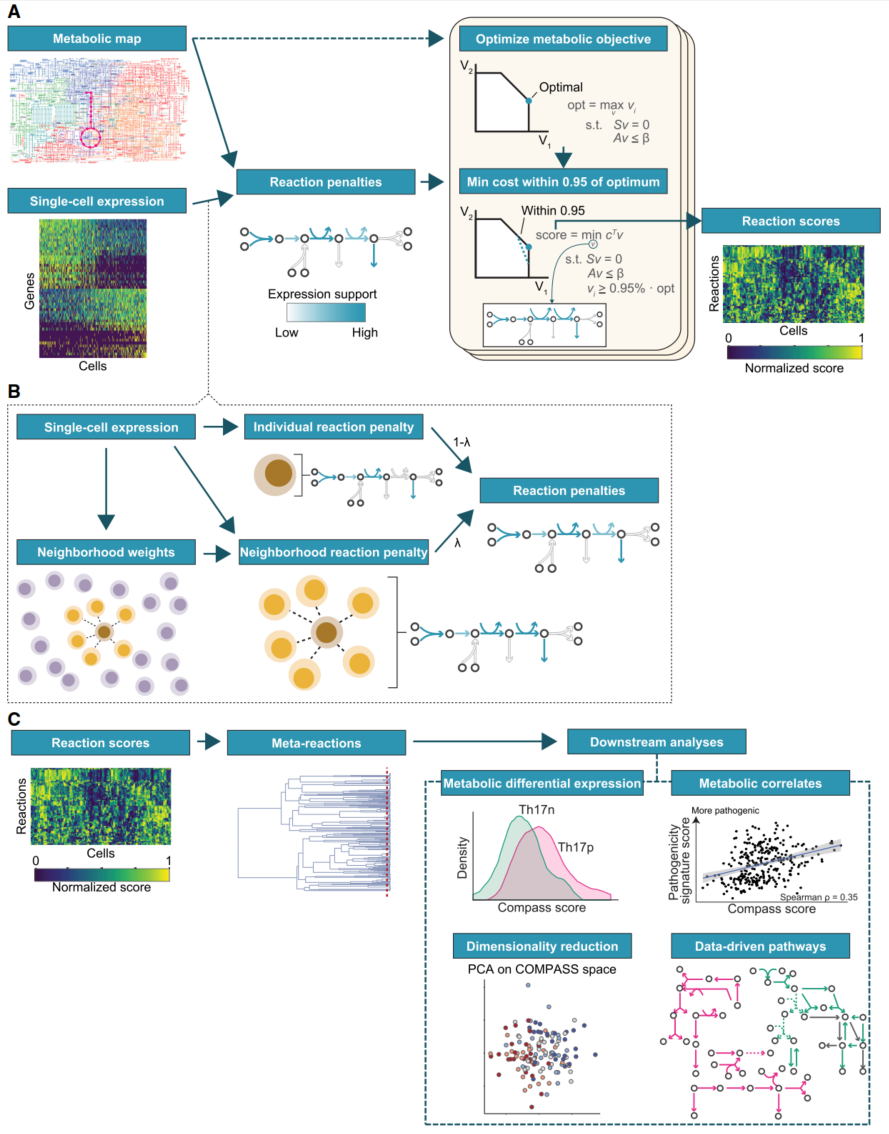
这篇文章的生物学重点是研究了Th17和Treg的代谢差异,为了得到不同细胞的代谢反应强度,他提出了一个Compass方法,简单来说,是把一个gene by cell matrix转换成一个reaction by cell matrix的方法。
化学反应数据库
首先,有一个已发表的化学反应database,叫Recon2,经过基本过滤,里面有7,440个化学反应,涉及2,626个代谢物。每个化学反应是一个配平方程式,也就是n个反应物生成m个生成物,是精准定量的。反应涉及一个或多个酶,而且可能是AND或OR的关系,可能有几个基因组成complex,或者几个基因的酶都可以催化同一个反应。
这篇文章把Recon2做成了一个EXCEL表,可以点开看看它具体的样子。
Compass算法
0. 流平衡分析
Compass 是FBA算法的一种,流平衡分析的思想是量化化学反应流,一些反应的生成物是另一些反应的反应物,每一种代谢物都有生成和消耗两个流向。流平衡的意思就是,任意时刻,对于任意代谢物,其生成和消耗都是相等的。当然生成和消耗也包括从细胞外摄入以及排出细胞,就是多了就排出去,少了就吸收进来。
每一个化学反应方程描述了生成一种代谢物需要消耗的原料、他们之间的比例、产生的副产物等。而反应$r$会有一个速率$v_r$,表示单位时间内该化学反应发生的摩尔量。流平衡分析的目的就是在约束所有代谢物都不产生积累或短缺的情况下,求出每个反应的反应速率$v$。
流平衡假设只是给解空间提出了一个约束,符合这个约束的解是无穷的,所以第二个假设就是,细胞是为了某个“目的”运行的,也就是要设置一个待优化的目标函数。一般来说,这个目标函数可以是细胞增大的速率(摄入总物质量-排出总物质量)。Compass会使用他自己的一个intuitive的目标函数。
1. 代谢反应模型
Compass使用了FBA里常用的Genome-scale metabolic model (GSMM),它有以下性质
- 化学计量矩阵$S$,行是代谢物,列是化学反应,矩阵值是在某个反应中某个代谢物的化学计量数,反应物是负数,生成物是整数
- 反应根据代谢物和酶所在的细胞区域分为几个compartment,分别是胞质、高尔基体、线粒体、细胞外基质
- 各区域之间的代谢物不共用,但一个区域的反应对另外区域的代谢物会有一个系数,来模拟物质的扩散
- 化学反应速率有上界和下界,它们是每个反应特异的
- 化学反应和基因之间存在关系,上面已经说过
流平衡约束就表示为
\[S \cdot v=\frac{d x}{d t}=0\]2. 算法
首先是符号约定

算法分3步
- 算出每个反应的最大流量

- 直观的理解是,满足流平衡假设(以及其它两个约束)的前提下,每个反应理论上可能获得的最大流量。如果其它反应都只为这个反应服务,这个反应单位时间能进行多少摩尔。
- 这一步是跟数据没有一点关系的,当$S$给定后,所有数据的$v_r^{opt}$都一样
- 算法主体,计算Compass score

- 这就复杂一点了,首先算了一个Penalty,这是
element-wise inversion of expression level of genes associated with reaction:scRAN-seq数据是个$g\times n$的矩阵,每个reaction有若干个关联基因,它们的和或均值(取决于AND还是OR)的倒数就构成这个$P_{m\times n}$矩阵 - 担心单细胞的sparsity,用它的k个近邻算了$P$矩阵的均值$P^N$,跟$P$加权平均
- 核心优化步骤,对每个细胞的每个反应,计算如果这个反应的速率达到其最大速率的95%,整体惩罚会是多少,所有反应的惩罚值之和作为Compass score
- 这就复杂一点了,首先算了一个Penalty,这是
- 对score的scaling处理

- 上一步得到的Compass-raw是越小表示反应越强,这一步把它转换成越大越强,并且变成相对值(减最小值)
点评
这是我自己的看法哈,不是作者写的
- Compass依赖一个IBM的商业软件Cplex,这是个用来解优化问题的C库,本来是学术用途免费的,但我试了几个国内大学的邮箱,都无法注册,要用的话只能下盗版或者让国外朋友帮忙了。卡脖子啊
- 虽然叫做GSMM,但Compass在求解的时候并没有把所有反应作为一个整体,而是为每个反应单独计算了“阻力分数”。在给不同反应算阻力的时候,其它反应的$v$值是不一样的,也就是说,这个方法并没有给整个系统求出一个最优的流量分配。我认为这是不可接受的,因为这样抛弃了作为FBA核心的系统性。
- 由于上面两个因素,Compass的计算性能和结果表现都堪忧,FBA系列的方法还需要在速度和整体性上有显著改进才会进入“能用”的级别
代谢物-受体介导的细胞通讯
文章标题是MEBOCOST: Metabolic Cell-Cell Communication Modeling by Single Cell Transcriptome,2022年5月31日刚挂上Biorxiv。作者是哈佛医学院、波士顿儿童医院的KAIFU CHEN。
这篇文章发现,包括Compass在内已经有好几个流平衡方法在做单细胞代谢推断了,那么就有两个问题,一是哪个方法做得更好,二是代谢推断做出来以后,能不能提升人们对细胞通讯的认知。
对于第一个问题,作者选了两个FBA方法来比较,Compass和scFEA,另外自己提了两个非常简单的模型,分别叫做算术平均法和几何平均法。做代谢推断的方法还有scFBA和一个gaussian mixture model,但前者是MATLAB平台的(商业软件),后者还没开源。算数平均法是将某个代谢物的生成反应涉及的所有基因求算术平均$P$,消耗反应涉及的所有基因求算数平均$S$,最终的分数$M=P-S$就是该代谢物的相对强度。几何平均法类似,只是平均数的算法换成几何平均。
验证用的数据则是来自CCLE project的928个cell lines,它们测了配对的bulk RNA-seq和metabolomics。每个代谢物只有二值化的有或无的标签。FBA类的方法天然并不提供代谢物浓度预测,而是用计算中的某些副产物来表征,对于Compass,是uptake reaction的强度,scFEA是balance result(scFEA没看,不知道是什么)。验证的指标则是看计算得到的metabolites present和实验的结果是不是相符,用Jaccard index。
结果非常的尴尬,Compass消耗了1000倍的计算资源,得到了比两种平均算法略差的结果,scFEA消耗了10倍的计算资源,得到了和平均算法差不多的结果(下图DE)。
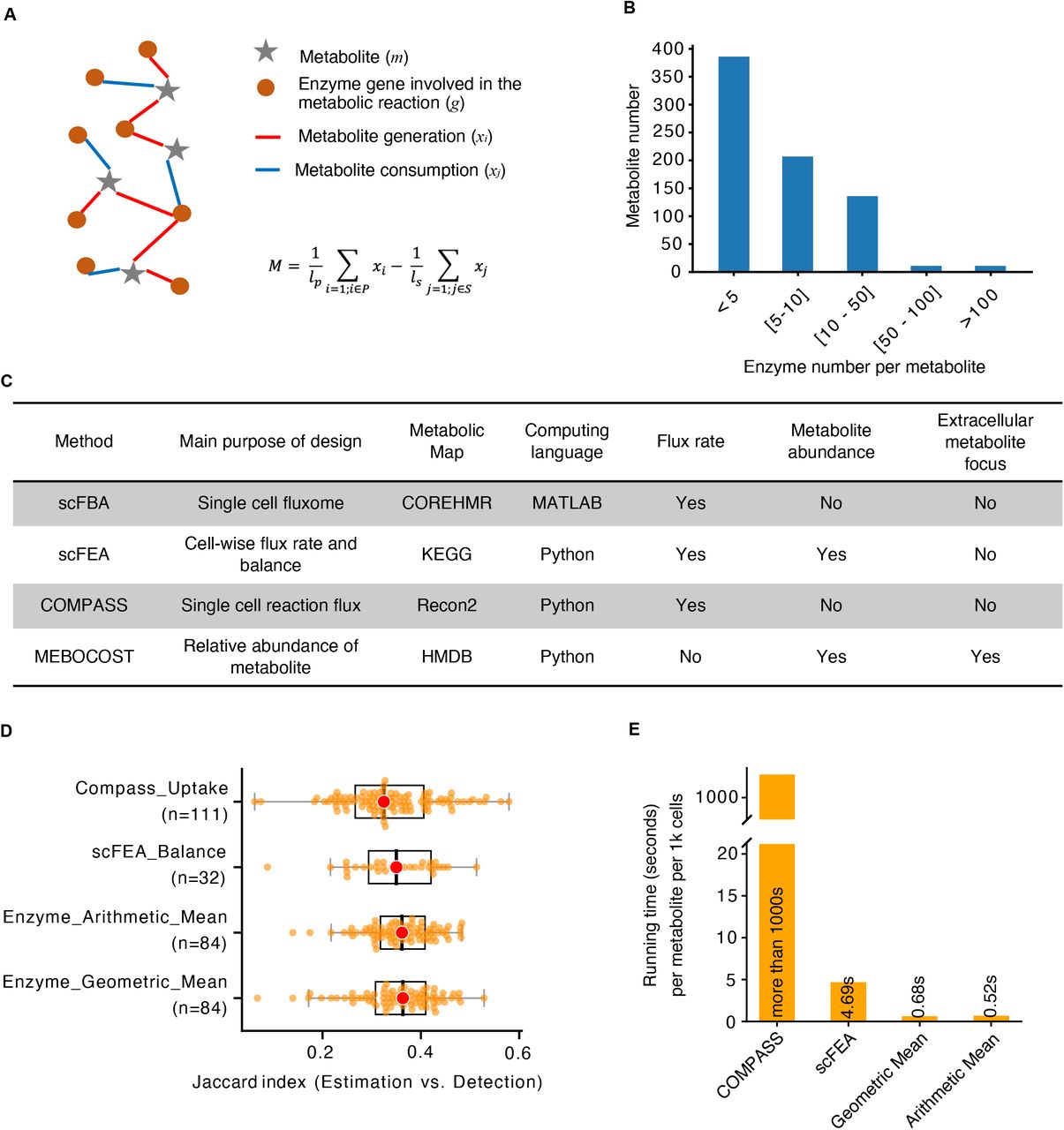
所以作者就决定用算术平均作为默认的计算metabolic score的方法。
接下来要做metabolic communication,为此还需要一个metabolite-sensor配对的数据库,其中sensor是一个蛋白,也就对应了一个基因。作者就自己造了一个数据库,通过糅合文献挖掘、Recon2、HMDB、GeneCards、GPCRdb、wikipedia,找到了440个配对。这些文件在github库的/MEBOCOST/data/mebocost_db/human里。
计算communication score就很简单了,对于某个sender cell type和某个receiver cell type,一堆metabolite-sensor的communication score就是metabolic present score和sensor expression level的乘积。p-value用1000次permutation做一个null distribution。
根据教程,MEBOCOST能画5种图,分别是event number barplot, circle network, dotplot, flow plot, violin plot。还有一个可以交互式画图的ipywidget。
点评
这篇文章所分析的是小鼠的棕色脂肪组织在遇到寒冷环境时,各细胞类型之间代谢通讯的变化,这个就是个添头,它的主要卖点还是MEBOCOST。
之前人们做的细胞互作分析,也可以叫ligand-receptor分析,是通过两个蛋白之间的交互来推断的。蛋白交互的情景在免疫系统里很常见,无论是chemokine介导免疫细胞的招募、interferon激活免疫反应、还是粘连蛋白辅助的细胞附着、免疫突触对靶标细胞的精确调控,都是蛋白和蛋白之间的交互。
但如果从整个人体的角度看,小分子-蛋白之间的交互才是细胞通讯的主流。大部分激素和神经递质都是脂质或有机酸这样的小分子,进一步说,信号转导、转录激活和抑制的调控、蛋白的修饰和泛素化降解等细胞内过程,里面也涉及大量的小分子。而生物体内的小分子除了葡萄糖等极少数可以直接从外界吸收,绝大部分都是代谢反应的结果,要搞清楚细胞内、细胞间发生了什么,是不可能只看蛋白而不看小分子的。
所以代谢通讯确实是个好方法,就现在的很粗糙的算相关基因平均值来预测代谢物含量的形式就已经能给出很多新的交互提示。但我认为以后随着FBA系列方法逐渐完善,多组学测序可以测到越来越多种类的代谢物,从系统出发一定是增加预测精度的正确方向。就现在而言,已经不妨把MEBOCOST加入scRNA-seq数据的标准分析流程。