In [2]:
%matplotlib inline
%load_ext autoreload
%autoreload 2
%load_ext line_profiler
In [3]:
import scanpy as sc
import random
import src.scanpy_unicoord as scu
import torch
from src.visualization import *
from line_profiler import LineProfiler
In [4]:
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_header()
# sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.settings.set_figure_params(vector_friendly=False)
load liver cancer data¶
In [5]:
adata = sc.read_h5ad(r'D:\hECA\Liver_cancer.pp.h5ad')
In [6]:
adata = adata.raw.to_adata()
sc.pp.normalize_total(adata, target_sum=1e4 ,exclude_highly_expressed= True)
sc.pp.log1p(adata)
adata
Out[6]:
model and training¶
In [7]:
scu.model_unicoord_in_adata(adata, n_cont=50, n_diff=0, n_clus = [],
obs_fitting=['Type'])
In [8]:
scu.train_unicoord_in_adata(adata, epochs=10, chunk_size=20000, slot = "cur")
In [9]:
fig = draw_loss_curves(adata.uns['unc_stuffs']['trainer'].losses)
# if save_figs:
# fig.savefig(os.path.join(savePath, 'img', 'fig1_lossCurves.png'))
fig.show()
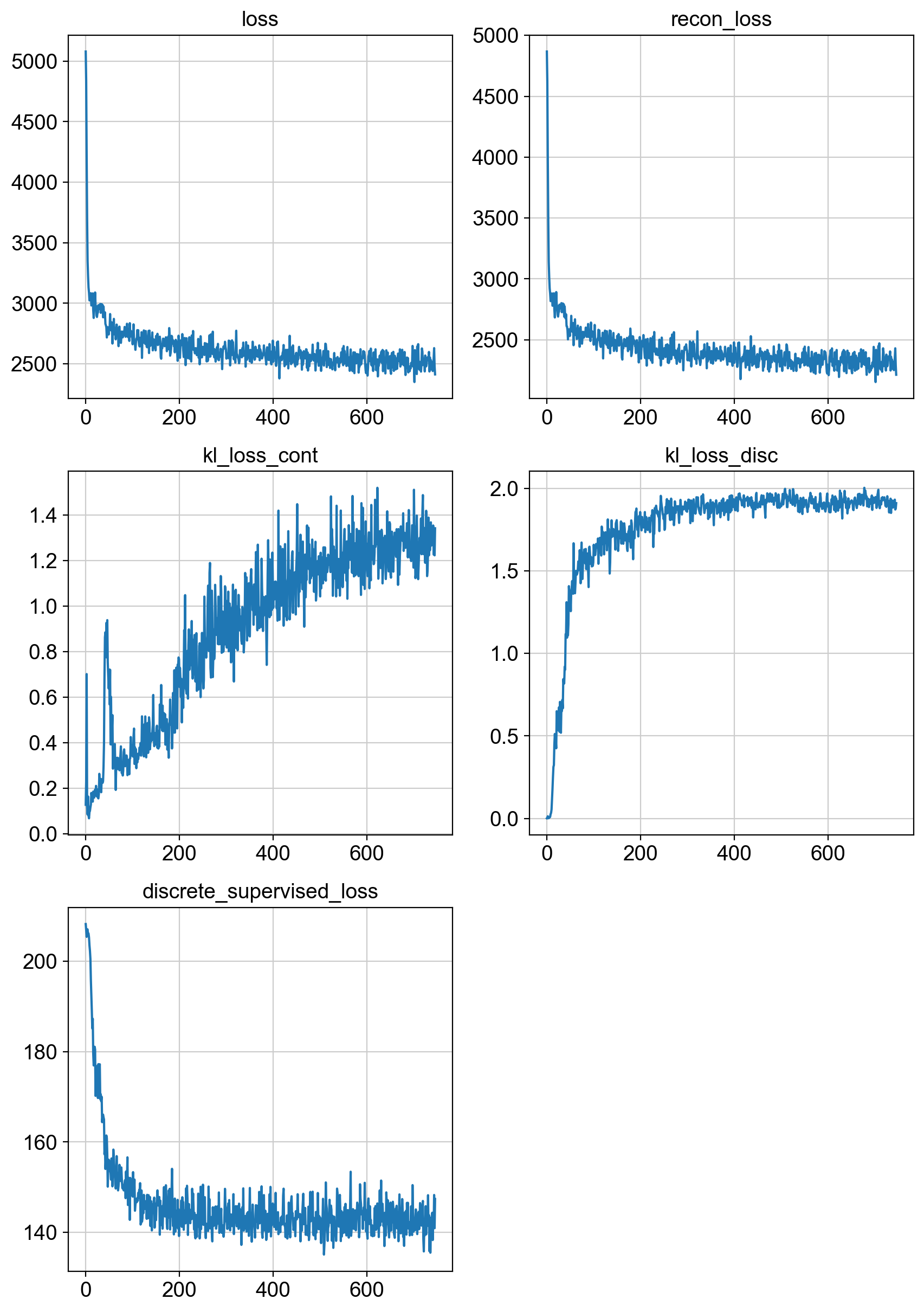
In [27]:
scu.embed_unicoord_in_adata(adata, chunk_size=5000)
In [28]:
sc.pp.neighbors(adata, use_rep='unicoord')
In [29]:
sc.tl.leiden(adata, resolution=0.5)
In [30]:
sc.tl.umap(adata)
In [1]:
sc.pl.embedding(adata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color=['leiden','Type','S_ID','Sample'], ncols=2)
In [43]:
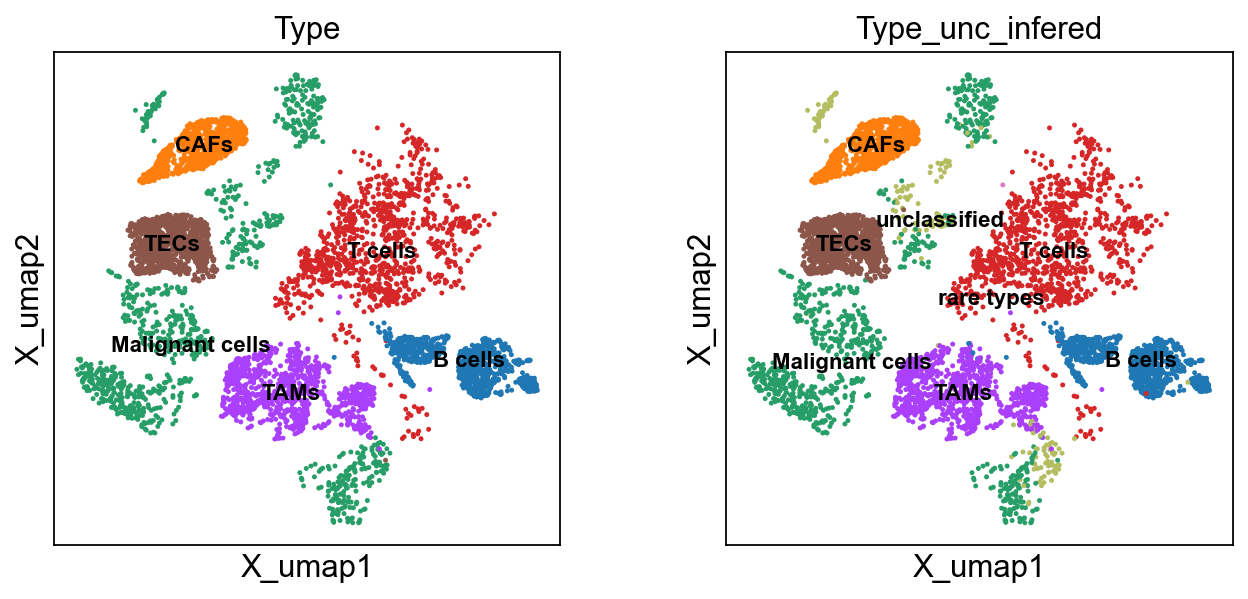
scu.predcit_unicoord_in_adata(adata)
In [44]:
ct1 = adata.obs.Type
ct2 = adata.obs.Type_unc_infered
ct1 = ct1[adata.obs.Type != 'unclassified']
ct2 = ct2[adata.obs.Type != 'unclassified']
accuracy_score(ct1, ct2)
Out[44]:
predict test set¶
In [32]:
bdata = adata[~adata.obs.unc_training,:].copy()
bdata
Out[32]:
In [33]:
scu.predcit_unicoord_in_adata(bdata, adata)
In [35]:
sc.pl.embedding(bdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['Type', 'Type_unc_infered'], ncols=2)

In [36]:
from sklearn.metrics import accuracy_score
In [45]:
ct1 = bdata.obs.Type
ct2 = bdata.obs.Type_unc_infered
ct1 = ct1[bdata.obs.Type != 'unclassified']
ct2 = ct2[bdata.obs.Type != 'unclassified']
accuracy_score(ct1, ct2)
Out[45]:
In [39]:
bdata.obs.groupby(['Type','Type_unc_infered']).size().unstack()
Out[39]:
generate data¶
In [10]:
import itertools
cells = list(itertools.chain(*[random.sample(list(adata.obs_names[adata.obs.Type==ct]), 1000) \
for ct in adata.obs.Type.value_counts().index if ct!='unclassified']))
In [11]:
bdata = adata[cells,:].copy()
bdata
Out[11]:
In [13]:
scu.predcit_unicoord_in_adata(bdata, adata)
In [14]:
sc.pl.embedding(bdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['Type', 'Type_unc_infered'], ncols=2)
gen without set cell type¶
In [56]:
cdata = scu.generate_unicoord_in_adata(bdata, adata)
In [57]:
sc.pp.normalize_total(cdata)
sc.pp.log1p(cdata)
sc.pp.highly_variable_genes(cdata)
In [58]:
cdata.raw = cdata
cdata = cdata[:, cdata.var.highly_variable]
cdata
Out[58]:
In [59]:
sc.pp.scale(cdata)
In [60]:
sc.tl.pca(cdata)
sc.pp.neighbors(cdata)
sc.tl.leiden(cdata)
In [61]:
sc.tl.umap(cdata)
In [62]:
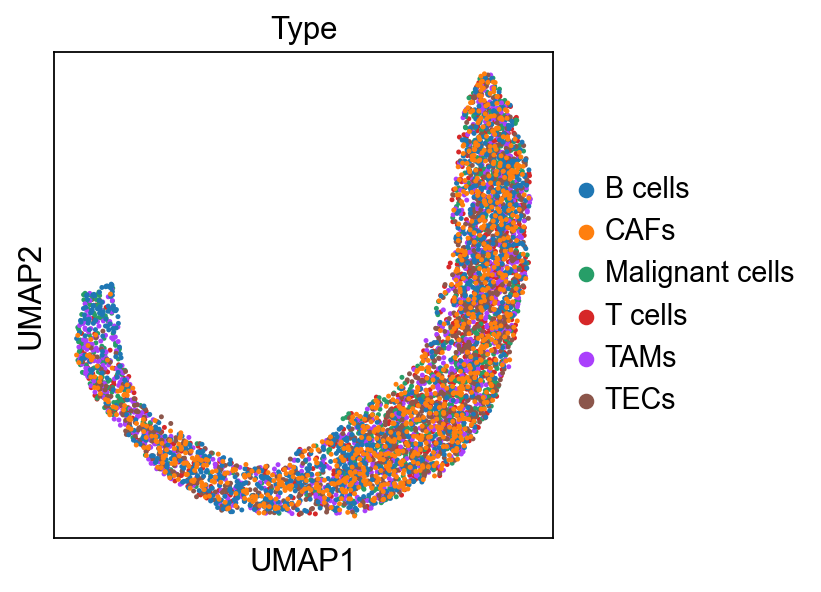
sc.pl.umap(cdata, color='Type')

In [63]:
cdata = cdata.raw.to_adata()
In [64]:
scu.predcit_unicoord_in_adata(cdata,adata)
In [67]:
sc.pl.umap(cdata, color=['Type','Type_unc_infered', 'CD3E','COL1A1'], ncols = 2)

gen and setting to T cells¶
In [68]:
cdata = scu.generate_unicoord_in_adata(bdata, adata,
set_value={'Type':'T cells'})
In [69]:
sc.pp.normalize_total(cdata)
sc.pp.log1p(cdata)
sc.pp.highly_variable_genes(cdata)
In [70]:
cdata.raw = cdata
cdata = cdata[:, cdata.var.highly_variable]
cdata
Out[70]:
In [71]:
sc.pp.scale(cdata)
In [72]:
sc.tl.pca(cdata)
sc.pp.neighbors(cdata)
sc.tl.leiden(cdata)
In [73]:
sc.tl.umap(cdata)
In [74]:
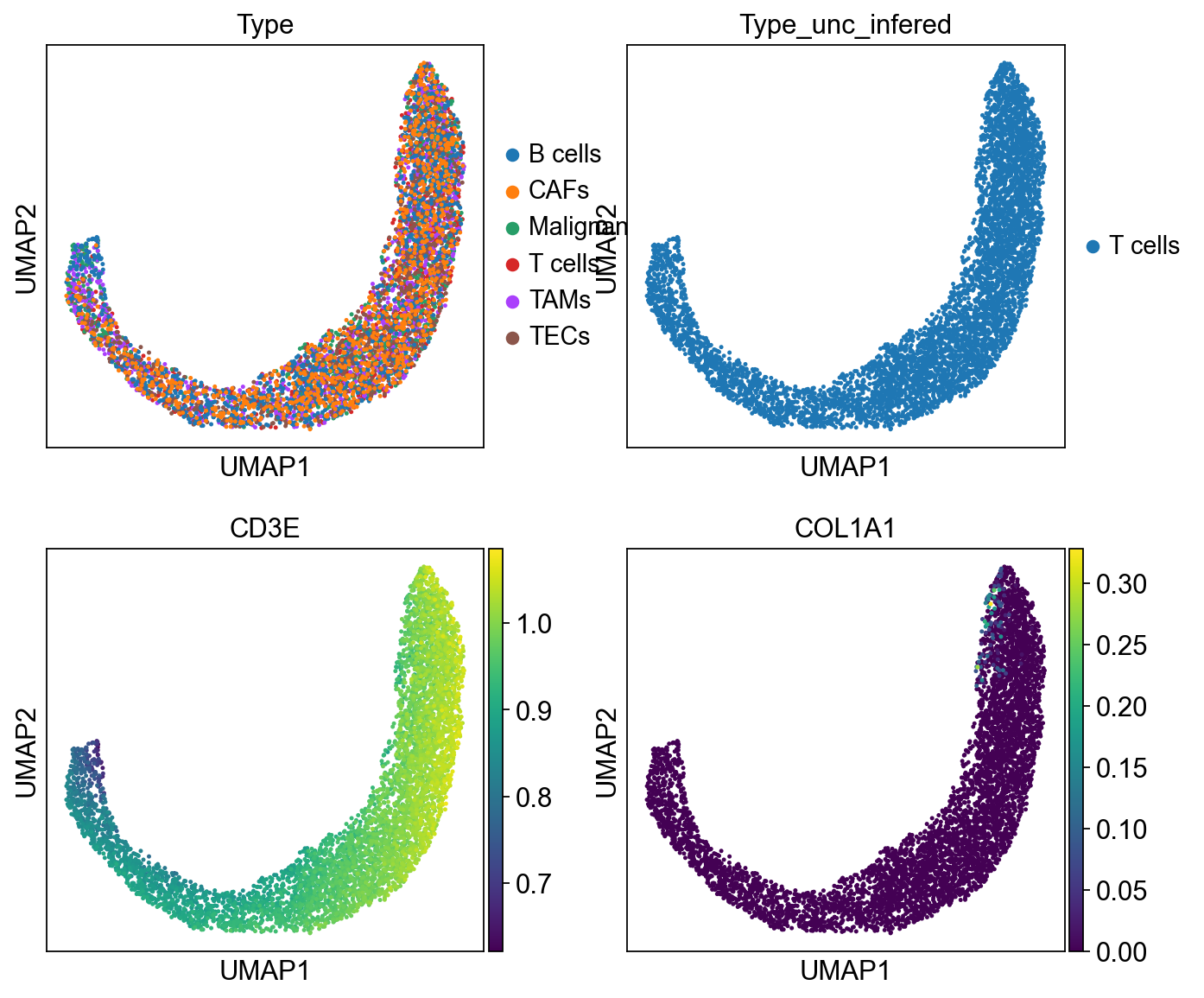
sc.pl.umap(cdata, color='Type')
In [75]:
cdata = cdata.raw.to_adata()
In [76]:
scu.predcit_unicoord_in_adata(cdata,adata)
In [77]:
sc.pl.umap(cdata, color=['Type','Type_unc_infered', 'CD3E','COL1A1'], ncols = 2)
gen from only T cells¶
In [87]:
bdata = adata[adata.obs.Type == 'T cells',:][:6000,:].copy()
bdata
Out[87]:
In [88]:
cdata = scu.generate_unicoord_in_adata(bdata, adata)
In [89]:
sc.pp.normalize_total(cdata)
sc.pp.log1p(cdata)
sc.pp.highly_variable_genes(cdata)
In [90]:
cdata.raw = cdata
cdata = cdata[:, cdata.var.highly_variable]
cdata
Out[90]:
In [91]:
sc.pp.scale(cdata)
In [92]:
sc.tl.pca(cdata)
sc.pp.neighbors(cdata)
sc.tl.leiden(cdata)
In [93]:
sc.tl.umap(cdata)
In [94]:
sc.pl.umap(cdata, color='Type')

In [95]:
cdata = cdata.raw.to_adata()
In [96]:
scu.predcit_unicoord_in_adata(cdata,adata)
In [97]:
sc.pl.umap(cdata, color=['Type','Type_unc_infered', 'CD3E','COL1A1'], ncols = 2)
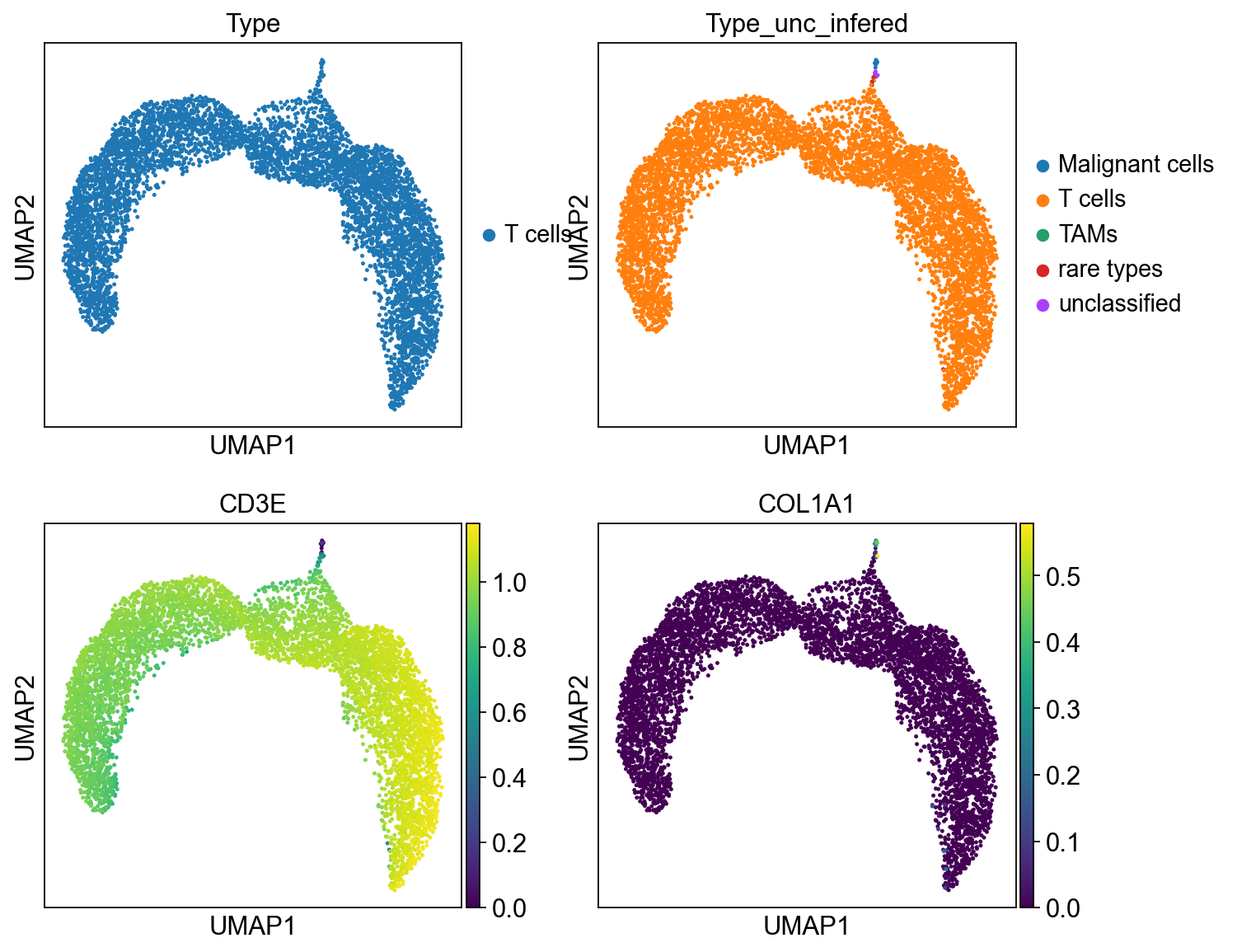
gen and setting to CAF cells¶
In [98]:
bdata = adata[cells,:].copy()
bdata
Out[98]:
In [99]:
cdata = scu.generate_unicoord_in_adata(bdata, adata,
set_value={'Type':'CAFs'})
In [100]:
sc.pp.normalize_total(cdata)
sc.pp.log1p(cdata)
sc.pp.highly_variable_genes(cdata)
In [101]:
cdata.raw = cdata
cdata = cdata[:, cdata.var.highly_variable]
cdata
Out[101]:
In [102]:
sc.pp.scale(cdata)
In [103]:
sc.tl.pca(cdata)
sc.pp.neighbors(cdata)
sc.tl.leiden(cdata)
In [104]:
sc.tl.umap(cdata)
In [105]:
sc.pl.umap(cdata, color='Type')
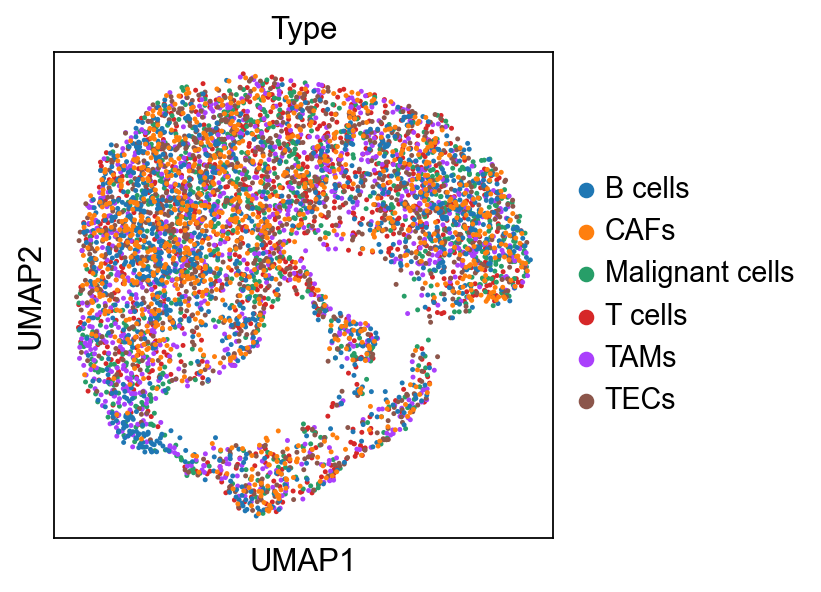
In [106]:
cdata = cdata.raw.to_adata()
In [107]:
scu.predcit_unicoord_in_adata(cdata,adata)
In [108]:
sc.pl.umap(cdata, color=['Type','Type_unc_infered', 'CD3E', 'COL1A1'], ncols = 2)
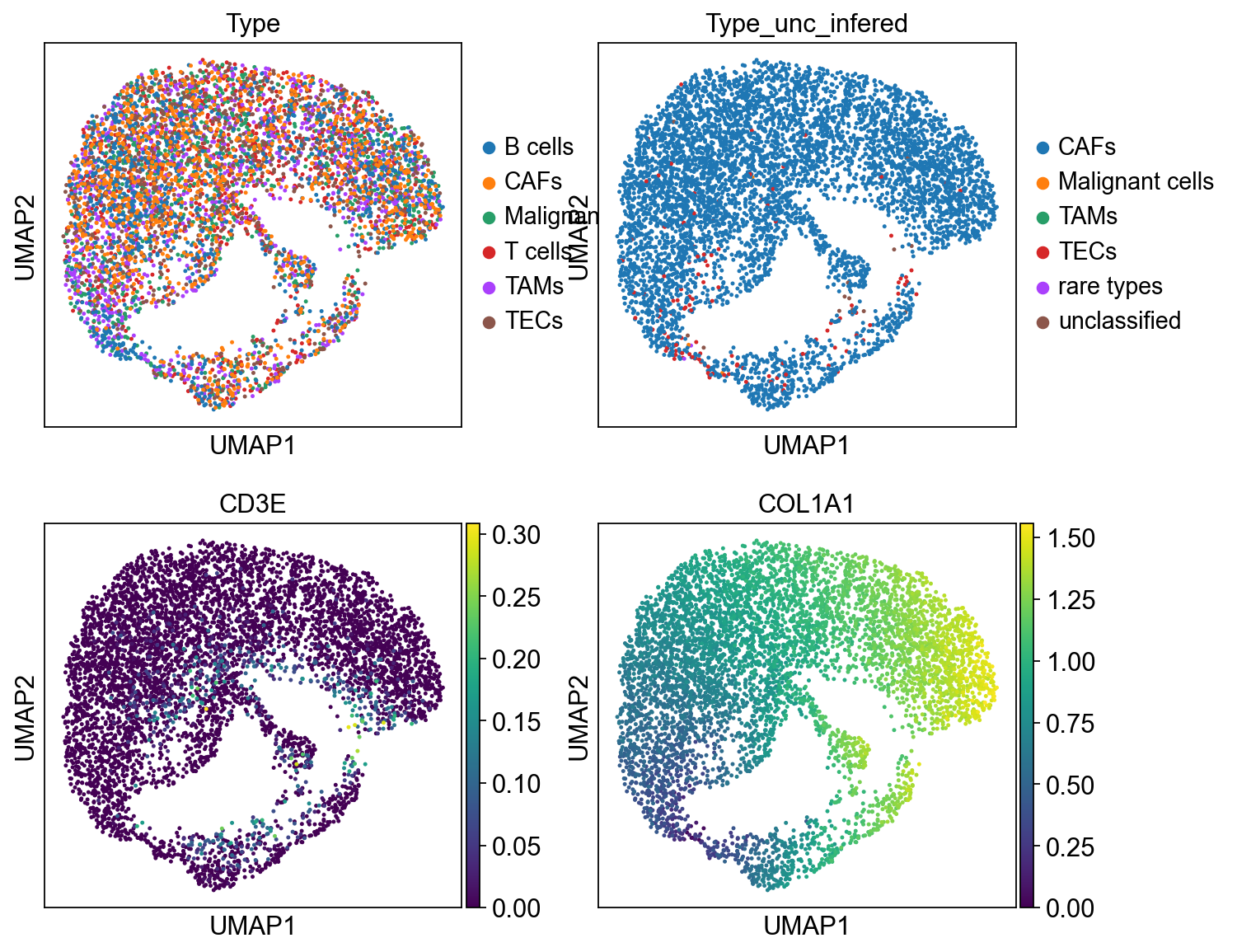
integrate gen and native data¶
gen & native¶
In [15]:
cdata = scu.generate_unicoord_in_adata(bdata,adata)
In [16]:
cdata
Out[16]:
In [17]:
ddata = cdata.concatenate(bdata, index_unique=None)
In [18]:
ddata
Out[18]:
In [19]:
sc.pp.normalize_total(ddata)
sc.pp.log1p(ddata)
sc.pp.highly_variable_genes(ddata)
In [20]:
ddata.raw = ddata
ddata = ddata[:, ddata.var.highly_variable]
ddata
Out[20]:
In [21]:
sc.pp.scale(ddata)
In [22]:
sc.tl.pca(ddata)
sc.pp.neighbors(ddata)
sc.tl.leiden(ddata)
In [23]:
sc.tl.umap(ddata)
In [25]:
sc.pl.umap(ddata, color=['Type','batch', 'CD3E', 'COL1A1'],
legend_loc='on data', ncols=2)

gen (T) & native¶
In [29]:
cdata = scu.generate_unicoord_in_adata(bdata,adata, set_value={'Type':'T cells'})
cdata
Out[29]:
In [31]:
ddata = cdata.concatenate(bdata, index_unique=None)
ddata
Out[31]:
In [32]:
sc.pp.normalize_total(ddata)
sc.pp.log1p(ddata)
sc.pp.highly_variable_genes(ddata)
In [33]:
ddata.raw = ddata
ddata = ddata[:, ddata.var.highly_variable]
ddata
Out[33]:
In [34]:
sc.pp.scale(ddata)
In [35]:
sc.tl.pca(ddata)
sc.pp.neighbors(ddata)
sc.tl.leiden(ddata)
In [36]:
sc.tl.umap(ddata)
In [37]:
sc.pl.umap(ddata, color=['Type','batch', 'CD3E', 'COL1A1'],
legend_loc='on data', ncols=2)
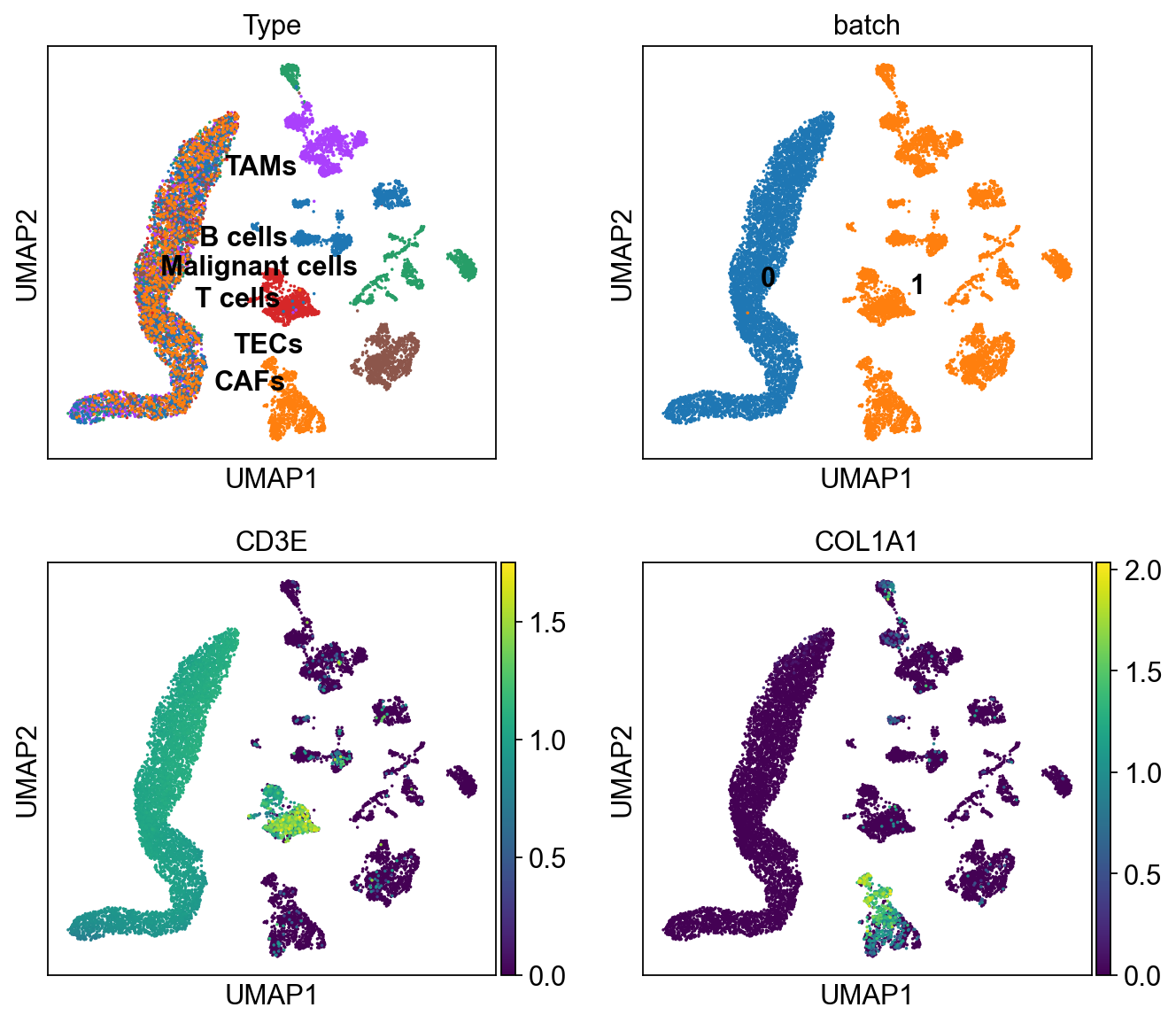
gen (CAF) & native¶
In [39]:
cdata = scu.generate_unicoord_in_adata(bdata,adata, set_value={'Type':'CAFs'})
cdata
Out[39]:
In [40]:
ddata = cdata.concatenate(bdata, index_unique=None)
ddata
Out[40]:
In [41]:
sc.pp.normalize_total(ddata)
sc.pp.log1p(ddata)
sc.pp.highly_variable_genes(ddata)
In [42]:
ddata.raw = ddata
ddata = ddata[:, ddata.var.highly_variable]
ddata
Out[42]:
In [43]:
sc.pp.scale(ddata)
In [44]:
sc.tl.pca(ddata)
sc.pp.neighbors(ddata)
sc.tl.leiden(ddata)
In [45]:
sc.tl.umap(ddata)
In [46]:
sc.pl.umap(ddata, color=['Type','batch', 'CD3E', 'COL1A1'],
legend_loc='on data', ncols=2)

gen (T) & gen¶
In [47]:
cdata = scu.generate_unicoord_in_adata(bdata,adata, set_value={'Type':'T cells'})
cdata
Out[47]:
In [48]:
cdata1 = scu.generate_unicoord_in_adata(bdata,adata)
cdata1
Out[48]:
In [49]:
ddata = cdata.concatenate(cdata1)
ddata
Out[49]:
In [50]:
sc.pp.normalize_total(ddata)
sc.pp.log1p(ddata)
sc.pp.highly_variable_genes(ddata)
In [51]:
ddata.raw = ddata
ddata = ddata[:, ddata.var.highly_variable]
ddata
Out[51]:
In [52]:
sc.pp.scale(ddata)
In [53]:
sc.tl.pca(ddata)
sc.pp.neighbors(ddata)
sc.tl.leiden(ddata)
In [54]:
sc.tl.umap(ddata)
In [55]:
sc.pl.umap(ddata, color=['Type','batch', 'CD3E', 'COL1A1'],
legend_loc='on data', ncols=2)
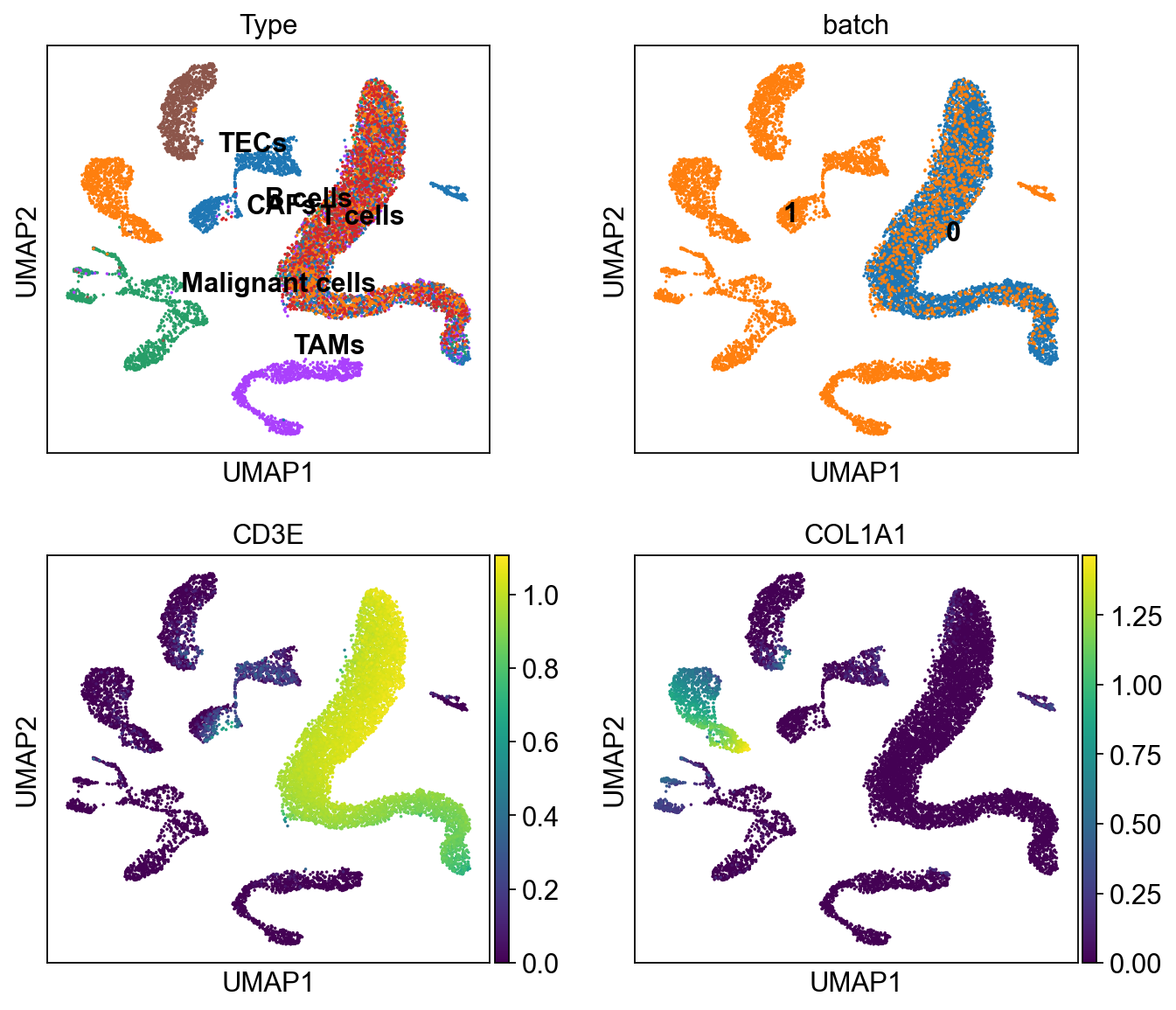
gen (CAFs) & gen¶
In [78]:
cdata = scu.generate_unicoord_in_adata(bdata,adata, set_value={'Type':'CAFs'})
cdata
Out[78]:
In [79]:
cdata1 = scu.generate_unicoord_in_adata(bdata,adata)
cdata1
Out[79]:
In [80]:
ddata = cdata.concatenate(cdata1)
ddata
Out[80]:
In [81]:
sc.pp.normalize_total(ddata)
sc.pp.log1p(ddata)
sc.pp.highly_variable_genes(ddata)
In [82]:
ddata.raw = ddata
ddata = ddata[:, ddata.var.highly_variable]
ddata
Out[82]:
In [83]:
sc.pp.scale(ddata)
In [84]:
sc.tl.pca(ddata)
sc.pp.neighbors(ddata)
sc.tl.leiden(ddata)
In [85]:
sc.tl.umap(ddata)
In [86]:
sc.pl.umap(ddata, color=['Type','batch', 'CD3E', 'COL1A1'],
legend_loc='on data', ncols=2)
