In [1]:
%matplotlib inline
%load_ext autoreload
%autoreload 2
%load_ext line_profiler
In [2]:
import scanpy as sc
import random
from unicoord import scu
from unicoord.visualization import draw_loss_curves
import torch
from line_profiler import LineProfiler
In [3]:
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_header()
# sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.settings.set_figure_params(vector_friendly=False)
load liver cancer data¶
In [4]:
adata = sc.read_h5ad(r'D:\hECA\Liver_cancer.pp.h5ad')
In [5]:
adata = adata.raw.to_adata()
sc.pp.normalize_total(adata, target_sum=1e4 ,exclude_highly_expressed= True)
sc.pp.log1p(adata)
adata
Out[5]:
model and training¶
In [6]:
scu.model_unicoord_in_adata(adata, n_diff=0,
obs_fitting=["S_ID", "Type"])
In [13]:
scu.train_unicoord_in_adata(adata, epochs=2, chunk_size=50000, slot = "cur")
In [16]:
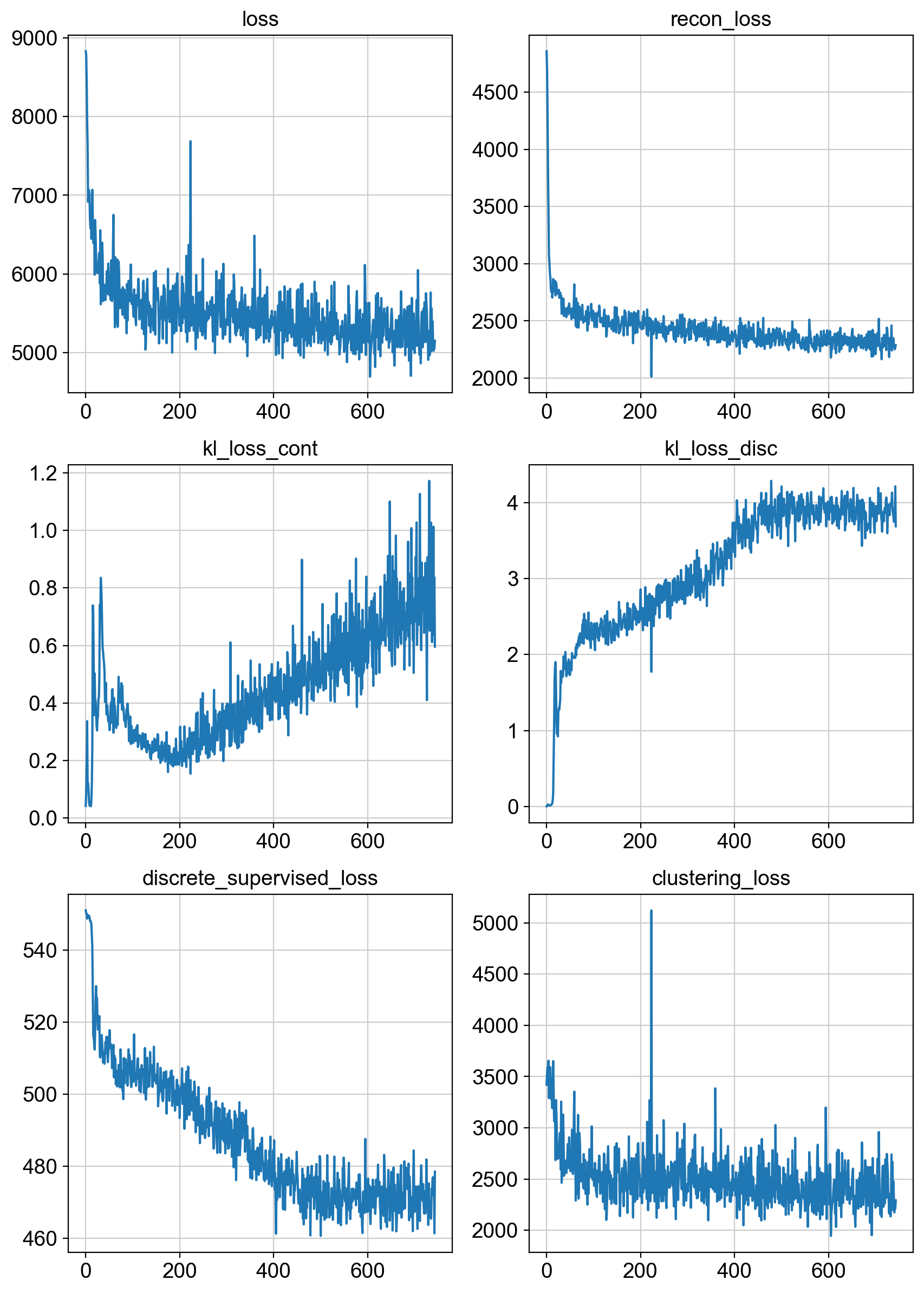
fig = draw_loss_curves(adata.uns['unc_stuffs']['trainer'].losses)
# if save_figs:
# fig.savefig(os.path.join(savePath, 'img', 'fig1_lossCurves.png'))
fig.show()
In [ ]:
for data in
In [11]:
scu.embed_unicoord_in_adata(adata, chunk_size=5000)
In [11]:
sc.pp.neighbors(adata, use_rep='unicoord')
In [12]:
sc.tl.leiden(adata, resolution=0.5)
In [13]:
sc.tl.umap(adata)
In [14]:
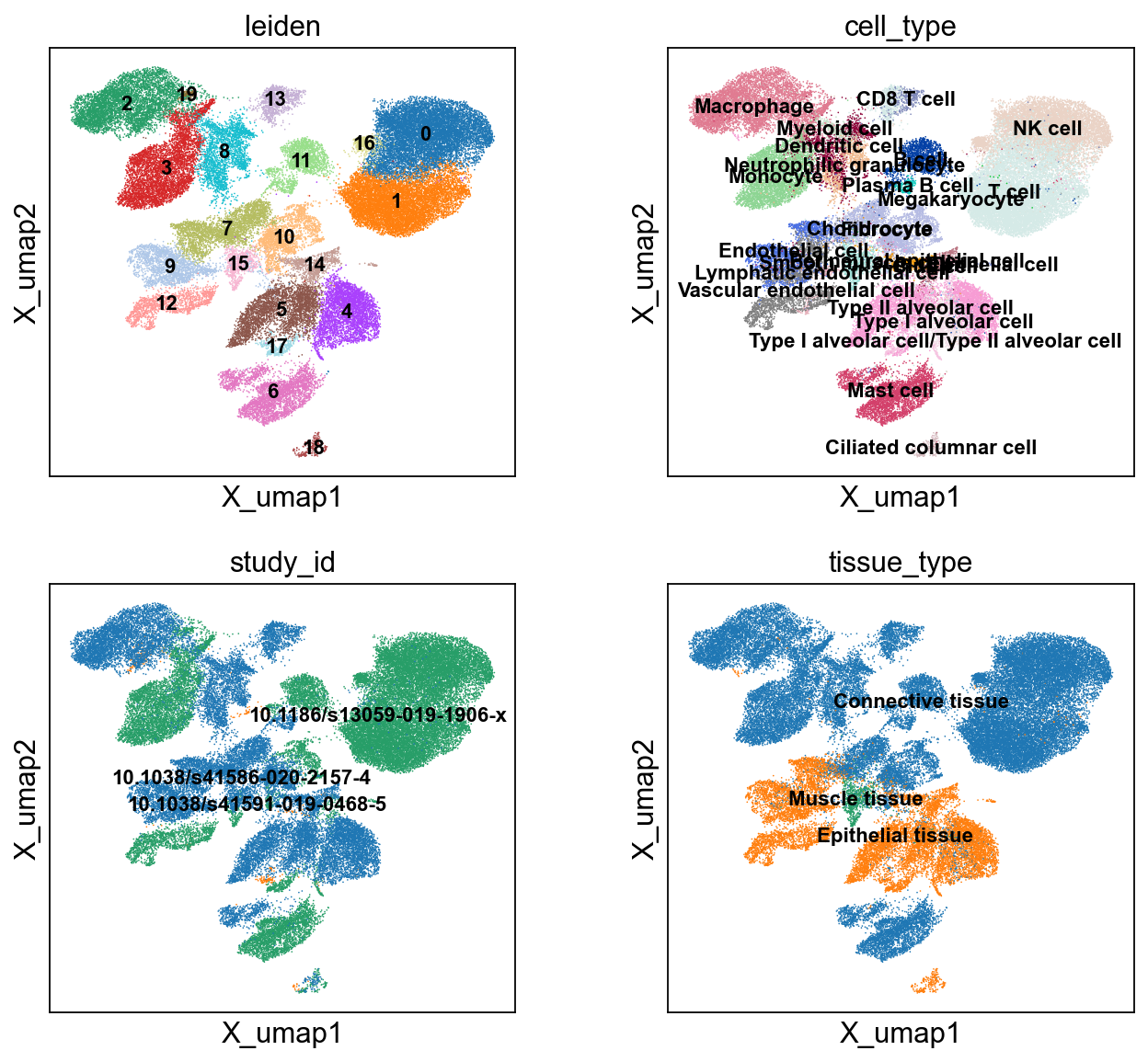
sc.pl.embedding(adata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color=['leiden','Type','S_ID','Sample'], ncols=2)

predict test set¶
In [30]:
bdata = adata[~adata.obs.unc_training,:].copy()
bdata
Out[30]:
In [31]:
scu.predcit_unicoord_in_adata(bdata, adata)
In [32]:
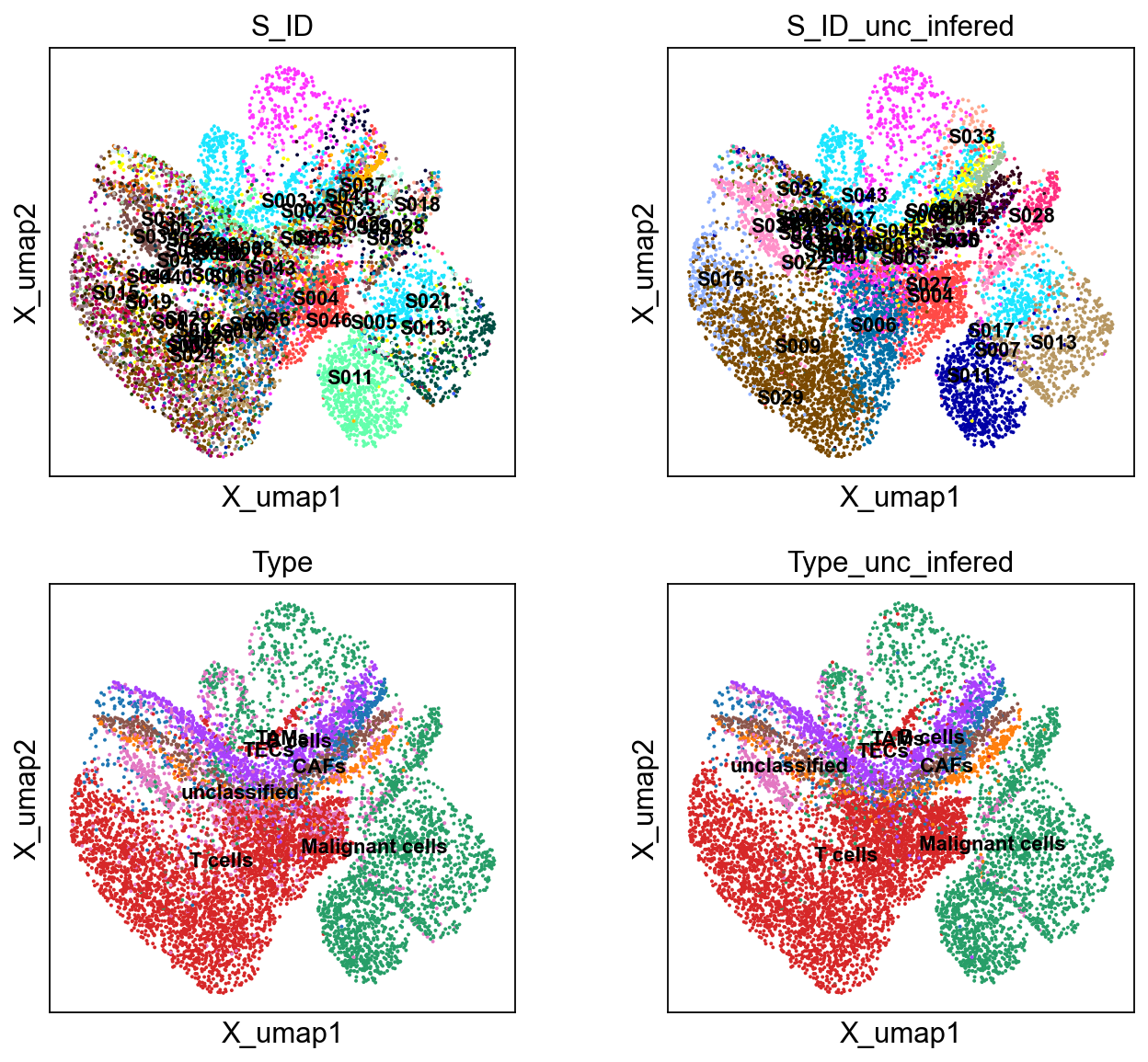
sc.pl.embedding(bdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['S_ID', 'S_ID_unc_infered', 'Type', 'Type_unc_infered'], ncols=2)
predict lung cancer data¶
In [24]:
cdata = sc.read_h5ad(r'D:\hECA\Lung_cancer_tLung.pp.h5ad')
cdata
Out[24]:
In [25]:
sc.pl.embedding(cdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color=['leiden','Cell_type.refined','Sample','Cell_subtype','Sample_Origin'], ncols=2)

In [26]:
cdata = cdata.raw.to_adata()
sc.pp.normalize_total(cdata, target_sum=1e4 ,exclude_highly_expressed= True)
sc.pp.log1p(cdata)
cdata
Out[26]:
In [27]:
scu.predcit_unicoord_in_adata(cdata, adata)
In [28]:
sc.pl.embedding(cdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['Sample', 'S_ID_unc_infered', 'Cell_type.refined', 'Type_unc_infered'], ncols=2)

predict lung ECA data¶
In [133]:
cdata = sc.read_h5ad(r'D:\hECA\Lung.Adult.pp.h5ad')
In [134]:
sc.pl.embedding(cdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
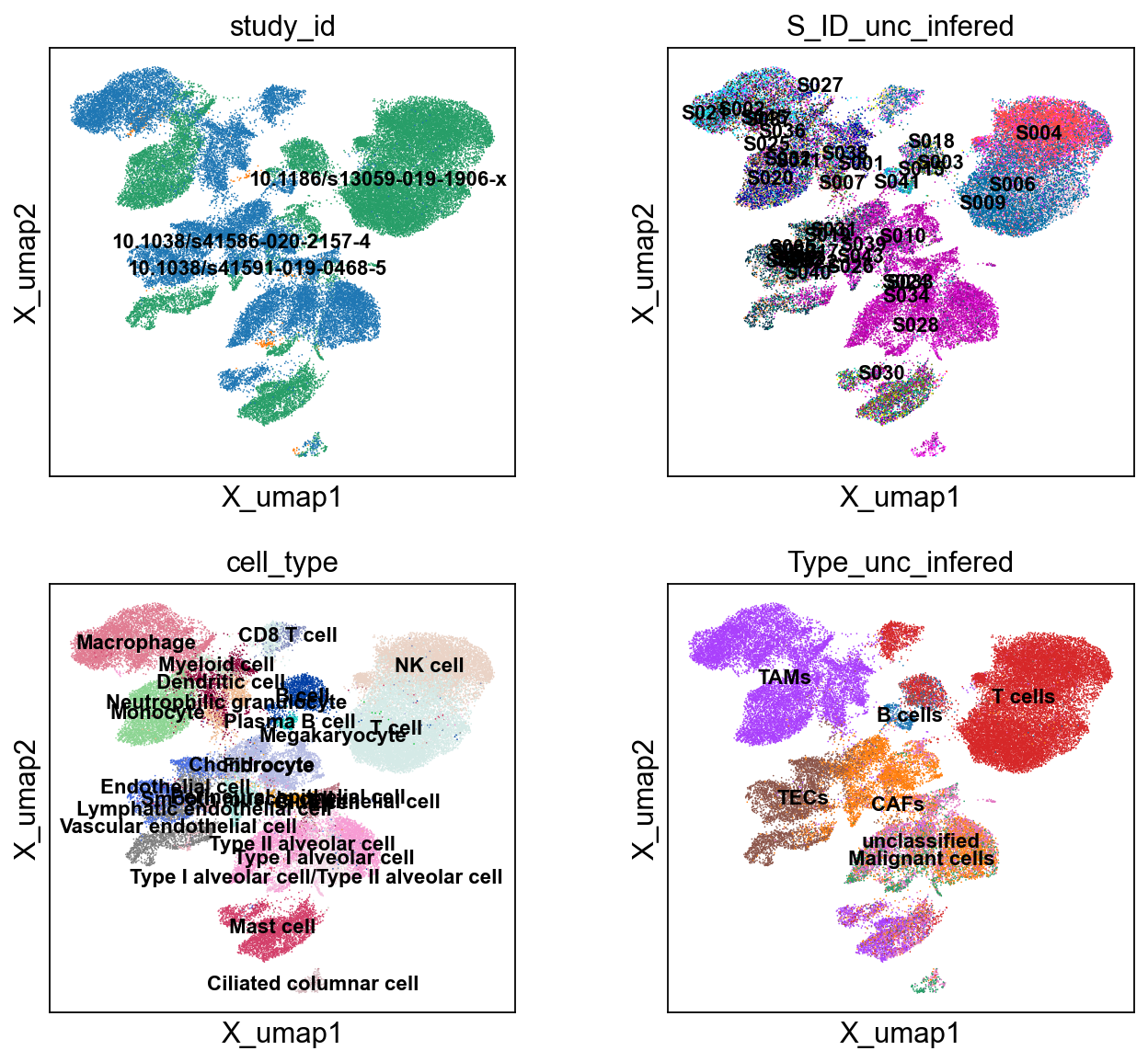
color=['leiden','cell_type','study_id','tissue_type'], ncols=2)
In [135]:
cdata = cdata.raw.to_adata()
sc.pp.normalize_total(cdata, target_sum=1e4 ,exclude_highly_expressed= True)
sc.pp.log1p(cdata)
cdata
Out[135]:
In [136]:
scu.predcit_unicoord_in_adata(cdata, adata)
In [137]:
sc.pl.embedding(cdata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['study_id', 'S_ID_unc_infered', 'cell_type', 'Type_unc_infered'], ncols=2)