In [1]:
%matplotlib inline
%load_ext autoreload
%autoreload 2
%load_ext line_profiler
In [2]:
import scanpy as sc
import random
from unicoord import scu
from unicoord.visualization import draw_loss_curves
import torch
from line_profiler import LineProfiler
In [3]:
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_header()
# sc.settings.set_figure_params(dpi=80, facecolor='white')
sc.settings.set_figure_params(vector_friendly=False)
Tabula Sapiens data¶
In [20]:
adata = sc.read_h5ad(r'F:\h5ad\tabularSapiens\obs_and_var.h5ad')
In [5]:
genes = pd.read_table('./protein_coding_genes.txt', header=None)[0]
g = list(set(genes) & set(adata.var_names))
In [6]:
adata
Out[6]:
In [7]:
scu.model_unicoord_in_adata(adata, genes_used=g,
n_cont=50, n_diff=0, n_clus = [],
obs_fitting=['cell_ontology_class','anatomical_information',
'method','organ_tissue','donor','gender','compartment'],
min_obs = 2000)
In [8]:
unc_stuffs = adata.uns['unc_stuffs']
In [9]:
for idx in range(3):
bdata = sc.read_h5ad(r'F:\h5ad\tabularSapiens\TBSP_%s.h5ad'%(str(idx)))
scu.train_unicoord_in_adata(bdata, unc_stuffs=unc_stuffs,
epochs=10, chunk_size=20000, slot = "cur")
torch.cuda.empty_cache()
In [10]:
fig = draw_loss_curves(unc_stuffs['loss'])
# if save_figs:
# fig.savefig(os.path.join(savePath, 'img', 'fig1_lossCurves.png'))
fig.show()
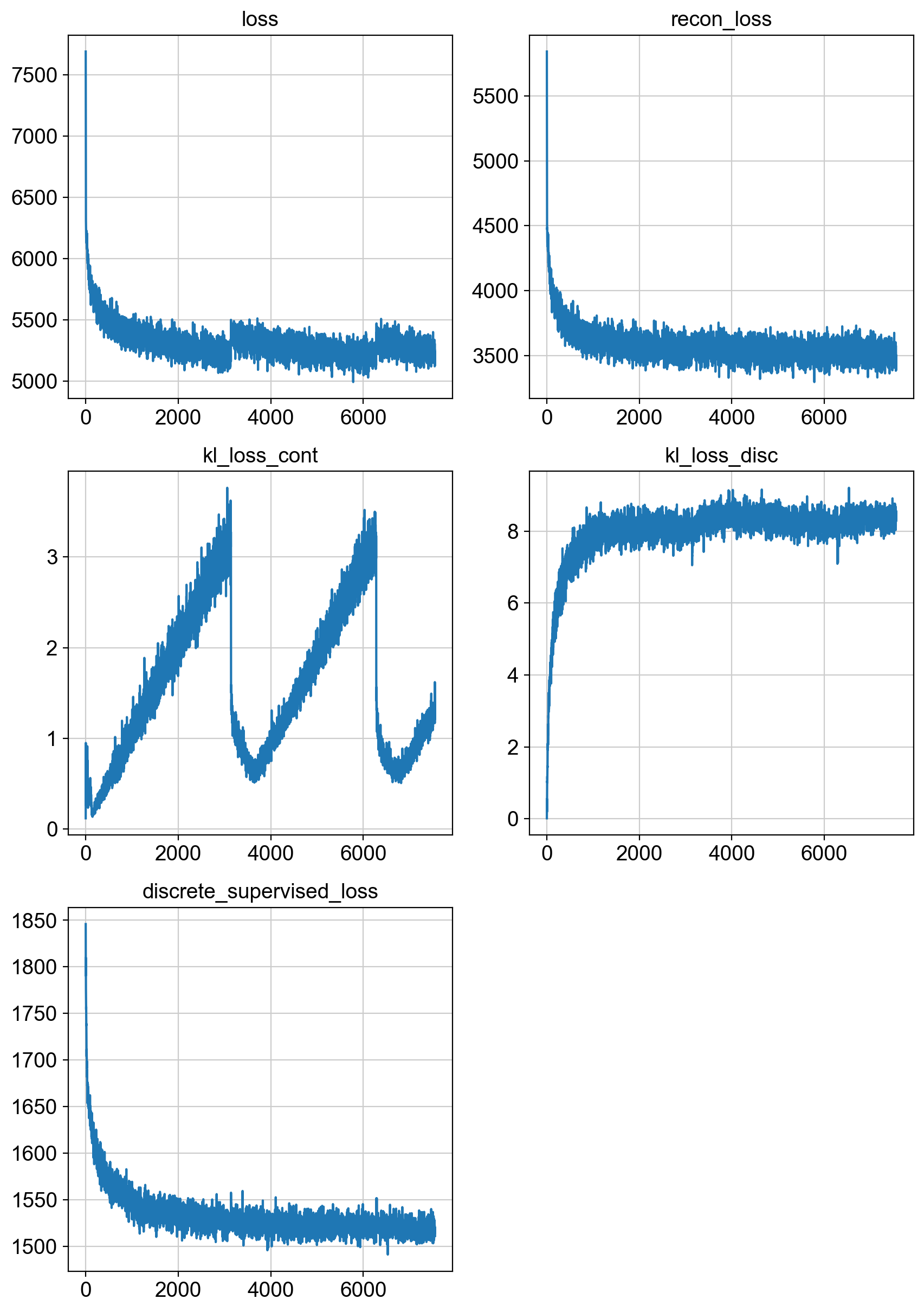
In [36]:
scu.write_scu_h5ad(adata, './pretrained_models/TBSP.h5ad', only_model=True)
predict liver cancer¶
In [4]:
cdata = scu.load_scu_h5ad('./pretrained_models/TBSP.h5ad')
cdata
Out[4]:
In [5]:
ddata = sc.read_h5ad(r'D:\hECA\Liver_cancer.pp.h5ad')
In [6]:
ddata = ddata.raw.to_adata()
sc.pp.normalize_total(ddata, target_sum=1e4 ,exclude_highly_expressed= True)
sc.pp.log1p(ddata)
ddata
Out[6]:
In [7]:
scu.predcit_unicoord_in_adata(ddata,adata_ref=cdata,chunk_size=1000)
In [8]:
sc.pl.embedding(ddata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['Type']+[s+'_unc_infered' for s in \
['cell_ontology_class','anatomical_information',
'method','organ_tissue','donor','gender','compartment']], ncols=2)

In [9]:
ddata.obs.groupby(['cell_ontology_class_unc_infered','Type']).size().unstack().to_csv('TBSP_HCC.csv')
predict LUAD¶
In [27]:
cdata = scu.load_scu_h5ad('./pretrained_models/TBSP.h5ad')
cdata
Out[27]:
In [28]:
ddata = sc.read_h5ad(r'D:\hECA\Lung_cancer_nLung.pp.h5ad')
In [29]:
ddata = ddata.raw.to_adata()
sc.pp.normalize_total(ddata, target_sum=1e4 ,exclude_highly_expressed= True)
sc.pp.log1p(ddata)
ddata
Out[29]:
In [31]:
scu.predcit_unicoord_in_adata(ddata,adata_ref=cdata,chunk_size=1000)
In [32]:
sc.pl.embedding(ddata, 'X_umap',legend_loc='on data', legend_fontsize=10,
color= ['Cell_type.refined','Cell_subtype']+[s+'_unc_infered' for s in \
['cell_ontology_class','anatomical_information',
'method','organ_tissue','donor','gender','compartment']], ncols=2)

In [12]:
ddata.obs.groupby(['cell_ontology_class_unc_infered','Cell_subtype']).size().unstack().to_csv('TBSP_LUAD.csv')
In [17]:
RP_genes = [g for g in cdata.uns['unc_stuffs']['genes_used'] if g.startswith("RPL") or g.startswith("RPS")]
In [18]:
pd.Series(RP_genes).to_csv('./RP_genes.csv')
In [26]:
ddata.obs.columns
Out[26]:
In [33]:
ddata.obs['Cell_subtype'][ddata.obs.organ_tissue_unc_infered == 'Muscle'].value_counts()
Out[33]:
In [34]:
ddata.obs.organ_tissue_unc_infered.value_counts()
Out[34]:
In [ ]:
adata.obs.organ_tissue.value_counts()